国产创新药迎来黄金发展期
国产创新药迎来黄金发展期今年是新中国成立70周年。在国庆前夕,动脉网特别策划“医疗创新系列报道”,通过我们对医疗各领域新技术新产品的报道梳理,试图还原医疗产业在国家倡导自主创新大背
今年是新中国成立70周年。在国庆前夕,动脉网特别策划“医疗创新系列报道”,通过我们对医疗各领域新技术新产品的报道梳理,试图还原医疗产业在国家倡导自主创新大背景下所取得的成就。
过去的五年是中国创新药蓬勃发展的五年,2018年药审中心批准了9个国产1类创新药,创历史新高。近年来批准的国产创新药中不乏代表性产品,如紧随世界步伐的三款国产PD-1单抗,中国首发全球新药罗沙司他,中国自主研发的First in Class药物本维莫德等。这些都是国内创新药企业不懈努力的关键成果。
除了创新药行业自身的努力,政策加资本打造的中国创新药新环境更是起到了关键性的辅助作用。
从国家政策层面,国家机关连续发文,为创新药产业发展打下政策基础;从资本市场方面,港股和科创板的接连开放为国内的创新药企业带来了更多的资金来源;从国际接轨方面,中国加入ICH直接将中国的创新药行业与国际串联,将国产创新药带到了更大的舞台上。
政策先行,三驾马车驱动创新药行业发展
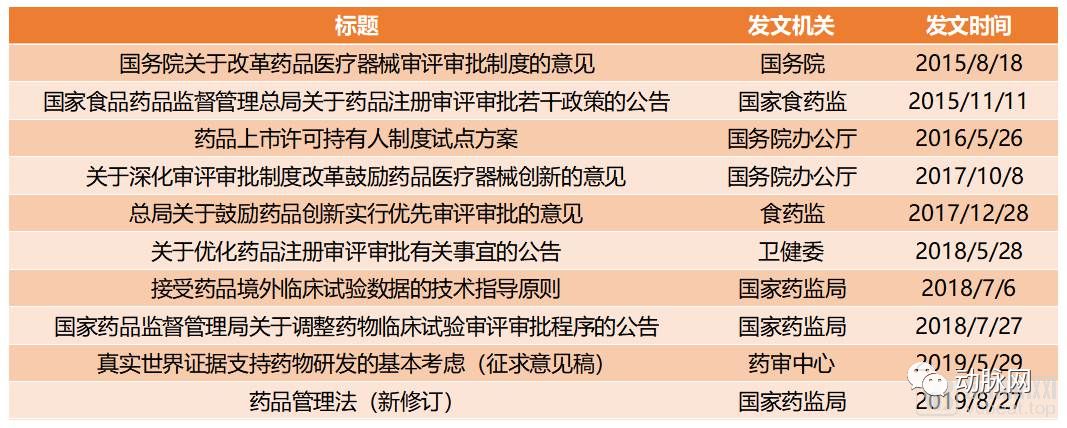
44号文以来国家有关促进创新药发展的主要政策
2015年8月,国务院印发的国发〔2015〕44号《关于改革药品医疗器械审评审批制度的意见》(以下简称44号文)正式拉开了国内创新药监管改革的大幕。44号文之后,厅字〔2017〕42号《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(以下简称42号文)再一次对44号文中提到的内容进行了强调,尤其突出了“促进药品创新和仿制药发展”、“改革临床试验管理”、“加快上市审评审批”、“加强药品医疗器械全生命周期管理”等几个要点。
如今回头看来,44号文和42号文在近几年的创新药相关政策中,起到了统领全局的作用。两份文件中提到的几大要点,都在经过几年的验证后,最终加入了2019年最新修订的《药品管理法》,成为正式的法律法规。
除了从概念上强调鼓励创新药行业发展之外,在近几年的医药新政中,临床审批加速、药品审评审批加速和药品上市许可持有人制度可以称得上的相关部门用实际行动促进行业发展的三驾马车。
1.临床审批加速:从18个月到2个月
在默示许可制度出台之前,从开始申报到最后拿到批件,临床试验审批一般要18个月左右。而这个时间,在澳大利亚是5天,美国、韩国是1个月,新加坡是1—2个月,欧盟是3个月,印度、俄罗斯是3—4个月。
更大的问题在于,这18个月的时间中,实际审批的时间大概就只有两个月,其余时间都花在各种不必要的流程中。报药审中心审批之前,先要报省局审核;报到药审中心之后还要排队,短则数月,长则一年;审评之后还不能直接上临床,药审中心的审评报告要转到注册司审查,注册司审查之后批件还要转到省局走内部程序,几经周转到了企业手上,又耽误了几个月时间。所以临床试验审批一直是我国创新药企业长期以来心头的痛。
44号文发布后,CFDA(现NMPA)积极响应,并迅速在2015年底出台了《国家食品药品监督管理总局关于药品注册审评审批若干政策的公告》。《公告》中将仿制药的生物等效性试验由审批制改为备案制,首先在仿制药上进行临床试验审批提速的尝试。
2018年7月27日,药审中心发布2018第50号文件《国家药品监督管理局关于调整药物临床试验审评审批程序的公告》(以下简称50号文)。50号文正式说明:自受理缴费之日起60日内,未收到药审中心否定或质疑意见的,申请人可以按照提交的方案开展临床试验。
不仅政策上跟进迅速,药审中心在执行上也毫不含糊。2018年11月5日,CDE官网主页热点栏目中,增加了“临床试验默示许可公示”一栏,并在当日公布了8个获默许的受理号。此时距50号文发布才刚刚过去了三个多月的时间。我国的临床试验申报程序正式进入默示许可时代。
默示许可从制度层面上大刀阔斧的砍掉了此前冗余的审批流程。药审中心在收到申报资料后5日内完成形式审查。符合要求或按照规定补正后符合要求的,发出受理通知书。再加上受理后的60天许可,一项申请材料齐全的临床试验,从申报到开展临床试验只需要65天的时间,比此前的审批速度加快了一年半。自默示许可公示栏开启后,已经有1040个临床试验以默示许可的方式通过审批。临床实验审批加速卓有成效。
2.上市审批加速:消化积压批件,创建优先审评机制
2015年9月,等待审评审批的药品注册申请达到了近22000件的高峰。国务院的44号文在这时看准时机,拍到了不断上升的曲线顶端。在政策推动下,国家食药监也在年底发布了更具体的政策指向,要求对八大类药品实现单独排队,加快审评审批。
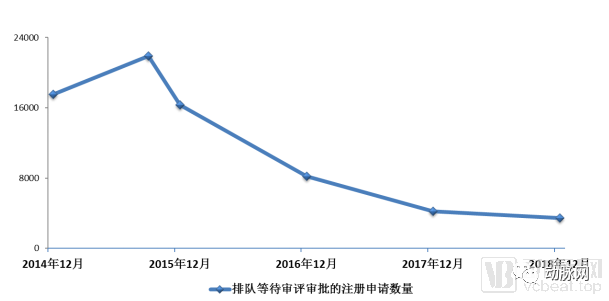
排队等待审评审批的注册申请数量变化情况
新政之下,药审中心开始加速消化此前积压的大量药品申请,到2017年12月,待审数量已经从22000锐减到4000件,初步完成了44号文中国务院提出的工作目标。
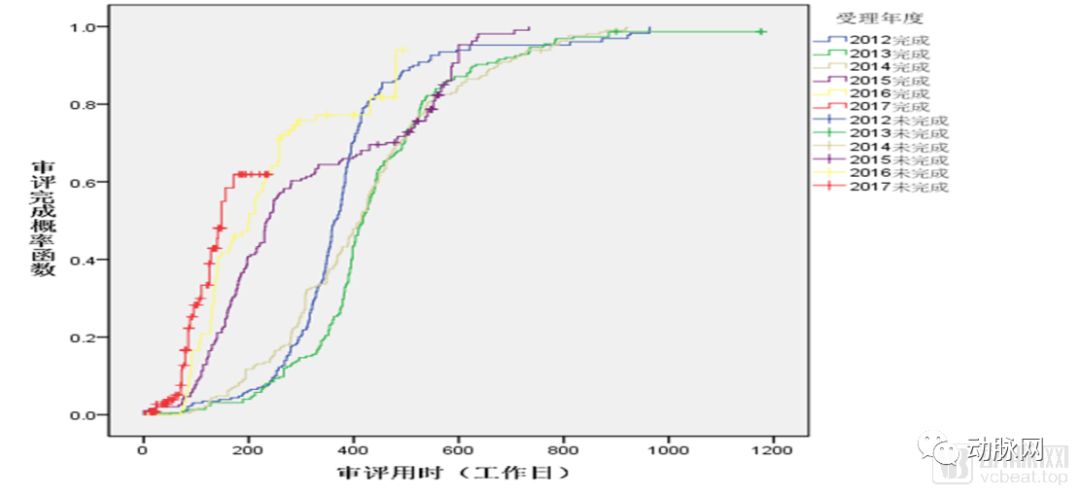
NDA审评用时逐年变化
同时,药品审批的平均时长也得到了极大的压缩。2017年的药品NDA审评时长中位数已经缩短到了180天左右,而这一数字在2013年是400天以上。
2017年12月,CFDA发布《总局关于鼓励药品创新实行优先审评审批的意见》,在其中再次对整体药品优先审评的落地方式进行了明确的说明,包括优先审评审批范围、优先审评审批程序和工作要求等落地过程中的关键问题,并对审评流程进行了明确的时间规定,保证优先审评审批工作高效有序进行。
临床试验审批加速和药品审评审批加速共同推动了药品全流程审批加速,让创新药不再苦于缓慢的审评审批效率。这对于本就研发投入大,耗时长的创新药行业来说,无疑是一针强心剂。加速审评还意味着创新药企业可以更多的开展国际多中心试验。以往因为国内审批缓慢造成的国内外时间差已经不复存在。
3.药品上市许可持有人制度:明确责任主体,鼓励技术创新
在2015年修订的《药品管理法》中,药品注册制度仍然是上市许可与生产许可相结合。也就是说,药品批准文号只颁发给具有《药品生产许可证》的生产企业。没有生产能力的企业和机构从基本条件上就无法获得药品批准文号,技术创新无法与最终市场收益直接挂钩,影响基础研发者们的积极性。
于是在2016年的5月26日,国务院办公厅发布《药品上市许可持有人制度试点方案》。《方案》要求在10个省(市)开展药品上市许可持有人制度试点,并对持有人的义务与责任、受托生产企业的义务与责任、及监管方式进行了全面的规定。
《方案》中提到,药品上市许可持有人可以“委托药品生产企业生产”,也可以“委托药品经营企业经营”。上市许可持有人只需要在药品的生产流通过程中进行监督,并保证药品的可追溯就可以了。这一制度彻底打破了我国此前研产销一体化的僵硬格局,让各个环节上的企业可以进行自由组合,各尽其职,提升药物创新效率。
药品上市持有人制度是我国医药产业发展的关键制度逻辑。第一,基础研发者们可以以持有人身份享有技术创新所带来的最终市场收益,有利于鼓励其积极研发;第二,明确了上市药品造成人身损害后的责任承担对象,促使持有人持续、主动关注上市药品的安全有效性;第三,赋予了持有人委托生产的权力,引导盘活闲置生产线、减少重复投产,提高我国医药制造业的产能利用率。
药品上市持有人制度的推行同时也催生了我国的CRO(合同定制研发)、CMO(合同定制生产)、CDMO(合同研发生产服务)等药品产业链上的特化角色。在药品生产流程中有专能的研发者可以在掌控核心竞争力的同时,借助CXO企业的力量进行研发、生产和药品上市,自己作为药品上市许可持有人最终获益。
3 首页 下一页 上一页 尾页-
合成生物行业2019年Q2融资报告:半年融资19亿美元,重点关注治疗领域2019-09-16
-
4+7新规:增至25省份和25种药品,这些区域的患者将受惠!2019-09-04
-
药品4+7推至全国,25省联盟集中采购文件出炉,25个品种待招采2019-09-02
-
当网售处方药合法、“假”药不假、GMP/GSP消失后,《药品管理法》带来的5大变化和14大要点2019-08-30
-
新版药品管理法:“新”面貌,“严”值很高2019-08-30
-
法无禁止即可为!网售处方药事实上开了“绿灯”!全面解读最新的药品管理法2019-08-27
-
新版药品管理法来了!这些新变化需要知道2019-08-27
-
全面解析新版医保目录对医药全行业的影响(下): 以治疗性药品为主导,中药注射剂无一新增!2019-08-27
-
2018-2019中国药品零售产业全景报告:传统药店形态将难以生存,产业进入转型期2019-08-16
-
80%的器械代表将失业,异常周密的器械招采方案,让对药品集采抱有的任何幻想都破灭了!2019-08-02
-
最新4+7药品集采调整的政策意图与实战2019-07-16
-
提高药品生产质量关键一环:制药设备管理工作尤为重要2019-06-25
-
3D打印药品助力制药领域实现新革命2019-06-18
-
药品回扣案件频发!两大冲击掀起“反贪风暴”2019-06-04
-
14省医药行业协会联名上书:建议国家医保局审慎推进新一轮药品集采试点2019-06-04