免疫细胞组分:immunedeconv包与Xcell批量处理文件
免疫细胞组分:immunedeconv包与Xcell批量处理文件一、immunedeconv包的下载与安装immunedeconv包:用于根据组织RNA测序数据估算免疫细胞组分的计
一、immunedeconv包的下载与安装
immunedeconv包:用于根据组织RNA测序数据估算免疫细胞组分的计算方法。该包的资源不同于一般的R包,并没有储存在CRAN或者bioconductor中。在github中查找immunedeconv,搜索页面出来的第一个就是可供R语言调用的immunedeconv包。点击进入便可以找到immunedeconv包的相关内容,以及下载、使用等相关信息。
点击进入,页面的最下方就是immunedeconv包下载的源代码,这是调用remote包中的install.github()函数进行下载。
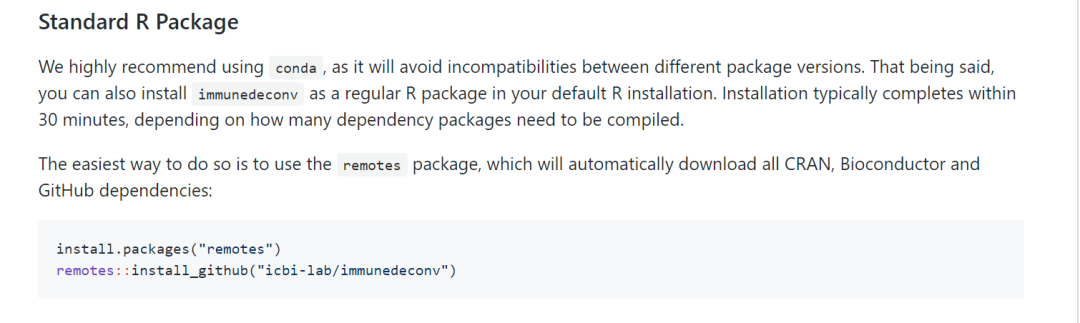
执行命令如下:
install.packages("remotes")
remotes::install_github("icbi-lab/immunedeconv"
网络差!以上代码执行失败,另寻他法。
尝试1:通过各种查阅资料,使用devtool包中的install.github(),依然报错了,使用remotes::install_github("icbi-lab/immunedeconv")和devtool::install.github("icbi-lab/immunedeconv"),都出现下面的错误:

尝试2:继续百度,找到一个类似的情况,"invalid multibyte string"提示此处存在错误编码字符。在这里附上原文标题,需要的朋友们可以参考一下,简书《[R|报错解决]Error in nchar(object, type = "chars") : invalid multibyte string, element 1》。
最终,多次努力之下如愿下载、安装、成功加载immunedeconv包(以下展示)。具体的过程就不再展示了,因为下载immunedeconv包需要的依赖包较多,而每个人的计算机已有的内置包不一样,所以每个人可能会遇到的一些问题不一样。总之,报错提示差什么就补什么。 执行命令如下:
#########安装immunedeconv##########
library(devtools)
Sys.setlocale(category = "LC_ALL",locale = "us") #调整内码格式
install_github("icbi-lab/immunedeconv")
library("immunedeconv")
ls("package:immunedeconv")
# [1] "dataset_racle"
# [2] "deconvolute"
# [3] "deconvolute_cibersort"
# [4] "deconvolute_epic"
# [5] "deconvolute_mcp_counter"
# [6] "deconvolute_quantiseq"
# [7] "deconvolute_quantiseq.default"
# [8] "deconvolute_timer"
# [9] "deconvolute_xcell"
# [10] "deconvolution_methods"
# [11] "eset_to_matrix"
# [12] "get_all_children"
# [13] "make_bulk_eset"
# [14] "make_random_bulk"
# [15] "map_cell_types"
# [16] "map_result_to_celltypes"
# [17] "scale_to_million"
# [18] "set_cibersort_binary"
# [19] "set_cibersort_mat"
# [20] "timer_available_cancers"
# [21] "xCell.data"
二、 deconvolute_xcell()的文件分析
deconvolute_xcell( ) 是immunedeconv包中的一种基于基因表达标志,用于评估混合组织中的64种免疫和基质细胞类型组成的计算方法。故首先用它来分析手头的已有bulk array基因表达矩阵。
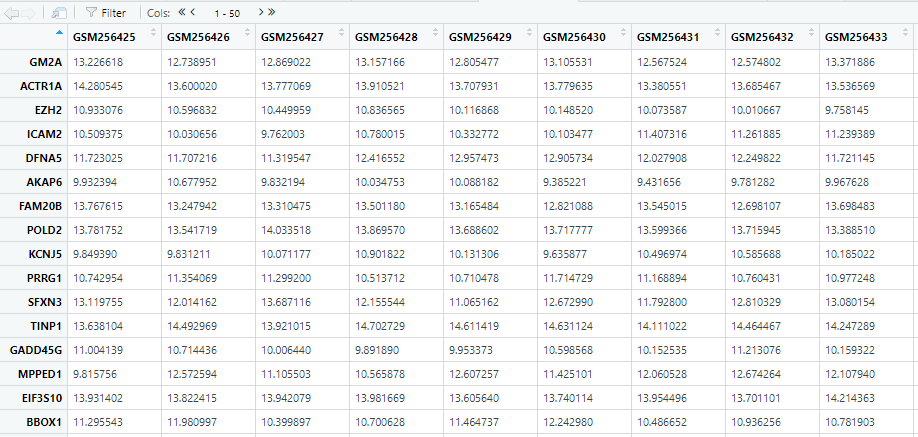
第一步:准备表达矩阵(行名已注释为gene symbol,与immunedeconv包中要求的hugo 基因名一致;列名为样品名)并写入R.
temptable<-read.table(file ="GSE10186_annotated_exprSet.txt" ,header = TRUE,sep = "",row.names = 1)
测试读取出内容如下:
第二步:执行deconvolute_xcell()并查看结果
abc <- deconvolute_xcell(gene_expression_matrix = temptable ,arrays = TRUE)
knitr::kable(abc[1:5,1:5], digits=2)
# | | GSM256425| GSM256426| GSM256427| GSM256428| GSM256429|
# |:----------|---------:|---------:|---------:|---------:|---------:|
# |aDC | 0.00| 0.03| 0.03| 0.04| 0.04|
# |Adipocytes | 0.07| 0.09| 0.09| 0.08| 0.10|
# |Astrocytes | 0.00| 0.00| 0.00| 0.00| 0.00|
# |B-cells | 0.02| 0.00| 0.00| 0.07| 0.00|
# |Basophils | 0.00| 0.13| 0.05| 0.06| 0.14|
第三步:将结果保存为.csv文件。
write.csv(file = "GSE10186_Xcell.csv")
三、for循环进行文件批量处理#
1.将所有要分析的同类型文件放在同一个文件夹下,将其设置为当前工作路径
#1.1获取该文件夹下的文件名和文件个数(我这里共有6个文件)
files<-dir()
files
# [1] "GSE10186_annotated_exprSet.txt" "GSE116174_annotated_exprSet.txt"
# [3] "GSE14520_GPL3921_annotated_exprSet.txt" "GSE14520_GPL571_annotated_exprSet.txt"
# [5] "GSE364_annotated_exprSet.txt" "GSE54236_annotated_exprSet.txt"
n <- length(files) #6L
#2. for循环语句
#2.1 简单试一试for循环是否可以正确执行。这里用的是批量提取文件的名称,并进行重命名
for ( i in 1:n){
print(paste(strsplit(files[i],"_")[[1]][1],"_",i,"_Xcell.csv"))
}
#2.2 正式进入循环:读入文件-执行deconvolute_xcell()-写出.csv文件,共循环6次。
for ( i in 1:n){
temptable<-read.table(file = files[i],header = TRUE,
sep = "",
row.names = 1,
quote = "")
deconvolute_xcell(gene_expression_matrix = temptable ,
arrays = TRUE) %>%
write.csv(file = paste(strsplit(files[i],"[_]")[[1]][1],"_",i,"_Xcell.csv"))
}
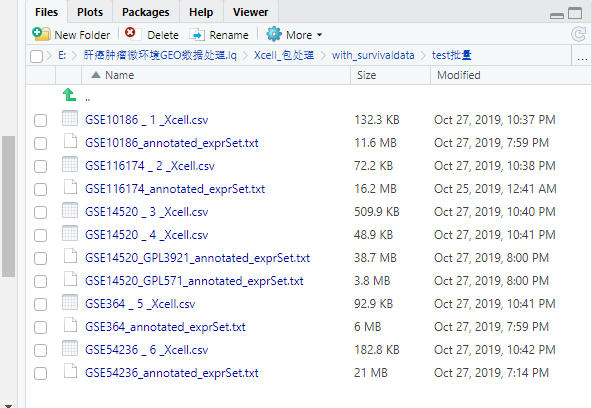
最终的结果:
四、批量处理过程中遇到的问题及解决方法
尽管自己认为for循环已经写得很完美了,但刚开始其实并没有想象中的那么顺利,囧…… 文件内容不变,还是刚开始的文件。但是系统一直报错:大意是结果只返回了第一条。在读文件的时候遇到了问题。具体什么原因,我就不得而知了,只有一步一步来尝试。
解决思路:
为什么结果只返回第一条?试一试循环,看是不是循环出问题了。可以看出,循环没有问题,因为可以输入6个结果;故可排除循环出问题的可能性。问题就出在读入文件-执行deconvolute_xcell()-写入.csv文件中。
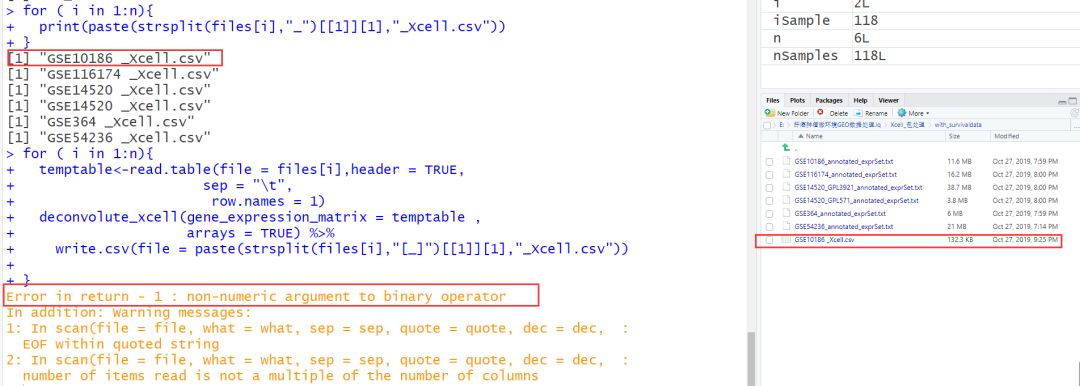
既然不能循环,那就只有拆分开来处理。如下:依次处理,依次看每一步的执行情况。 第一个顺利。
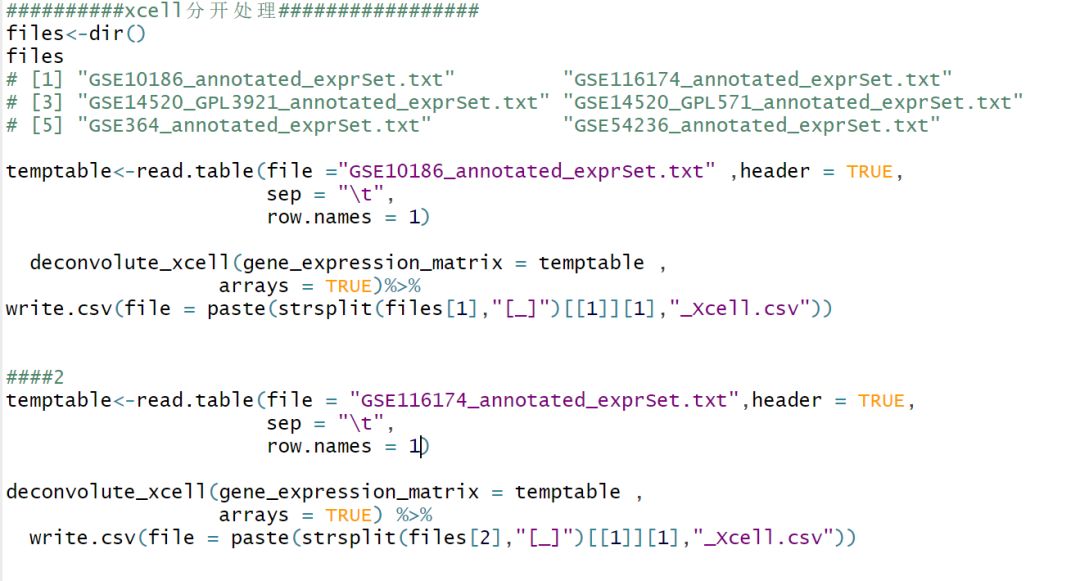
oh......第二个好像出问题了?原来for循环的问题出现在这里。

打开原文件查看,肉眼并不能发现数据格式问题。既然它继续提示字符带有引号(尽管在数据处理之前我已经统一去除了引号,这里为什么会这样提示,我也不知道原因),那就去掉引号吧。 read.table()中quote=""参数可以用于指定包围字符型数据的字符。

增加该参数后,再次执行第二个文件的数据处理,结果就很顺利。在后面的4个文件中,也使用该参数,鉴定完毕,其他几个文件没有问题。
最后再次尝试for循环:在for循环中添加该参数,再次执行for循环。结果不出意料,非常顺利。就是我上面展示的“2.2 正式进入循环:读入文件-执行deconvolute_xcell()-写出.csv文件,共循环6次”后的结果了。
结论:在执行for循环的时候,例如从第1个文件到第20个文件,如果在第5个文件处出错,那么系统就可以执行到第4个文件,然后提示返回第1个文件,故我们需要解决第5个文件的问题。唯有解决第5个文件的问题,或者跳过第5个文件,才可以继续执行第6个文件到第20个文件的命令。
-
第四批国采文件解读发布,2月3日报价见!2021-01-28
-
富士康开始正式进军汽车行业,郑显聪上任电动汽车平台首席执行官2021-01-18
-
工业机器人末端执行器分类2020-12-29
-
极飞科技:从感知、执行出发,布局无人化农业生态系统2020-12-23
-
爱普生研发设计全球首个*1 PaperLab干纤维纸张循环系统2020-12-07
-
贾跃亭再成被执行人:破产依然被执行2020-12-07
-
医保信息系统推进执行,灰色收入全部出局!2020-12-02
-
工业环境保护、循环经济等方面,宝马做到了可持续吗?2020-11-20
-
美商务部决定暂不执行TikTok禁令 态度缓和?2020-11-13
-
华为“懂行大会2020”:打造“懂行”价值循环,共赢行业新价值!2020-11-06
-
华为蔡英华:以“懂行”的价值循环,创造更大的行业价值2020-11-06
-
智慧生态创新峰会成功举办 聚焦数字化深度打造高质量“双循环”2020-11-02
-
国内国际“双循环”开创中医药发展新格局2020-10-31
-
聚材道的“生态小循环”,如何撬动城市大循环?2020-10-31
-
滴滴网约车执行总裁陈熙将离职 孙枢接任2020-10-21